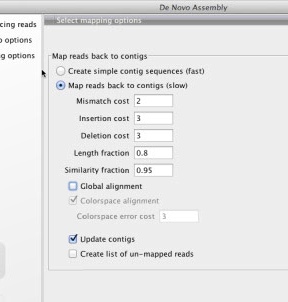
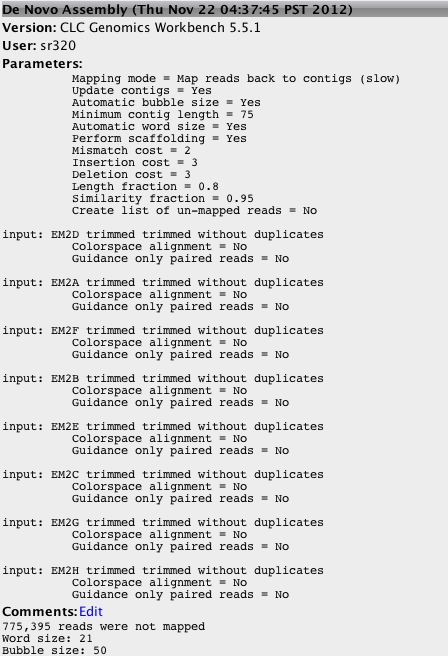
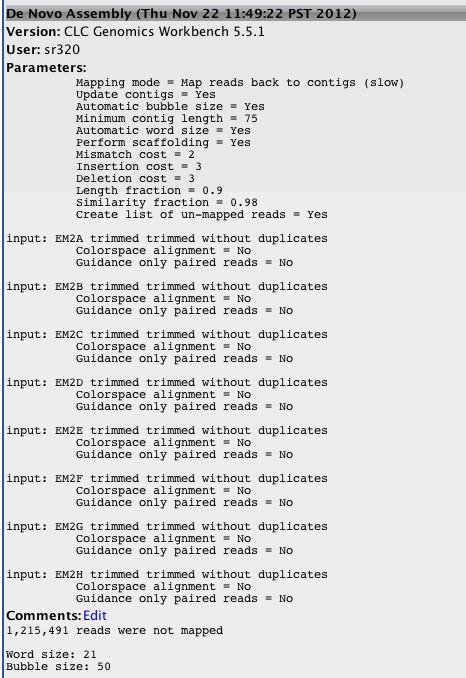
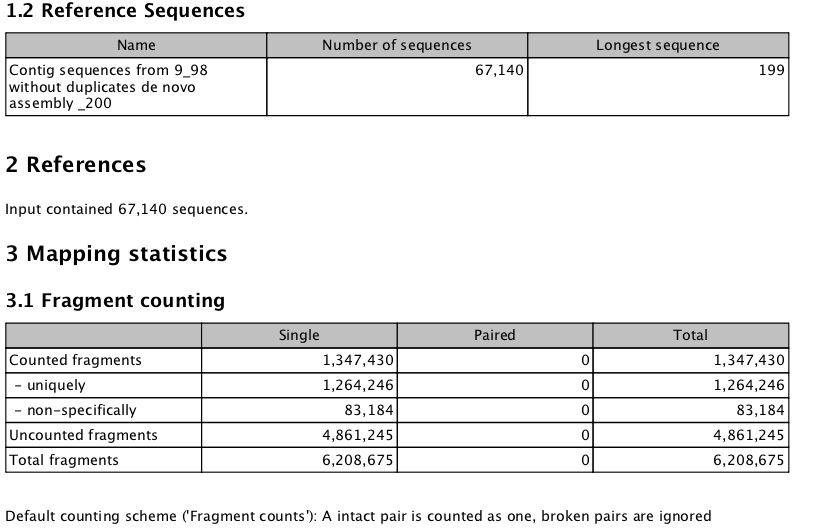
- taking trim trim (remove 4bp 5' and <90bp length)
and "Removing Duplicates"
De novo assembly of 8 libraries without duplicates
--
SAM format
Fasta of contigs
--
Assembly with different parameters (stricter)
---
subset of denovo
for denovo smaller than 200 bp - (went form 72k to 67k)
note that this is assembly was done with duplicates removed, thus singlets (those that did not assemble) could be important
------
Running RNA-seq on this backbone